Our research: decoding how phages and bacteria co-evolve
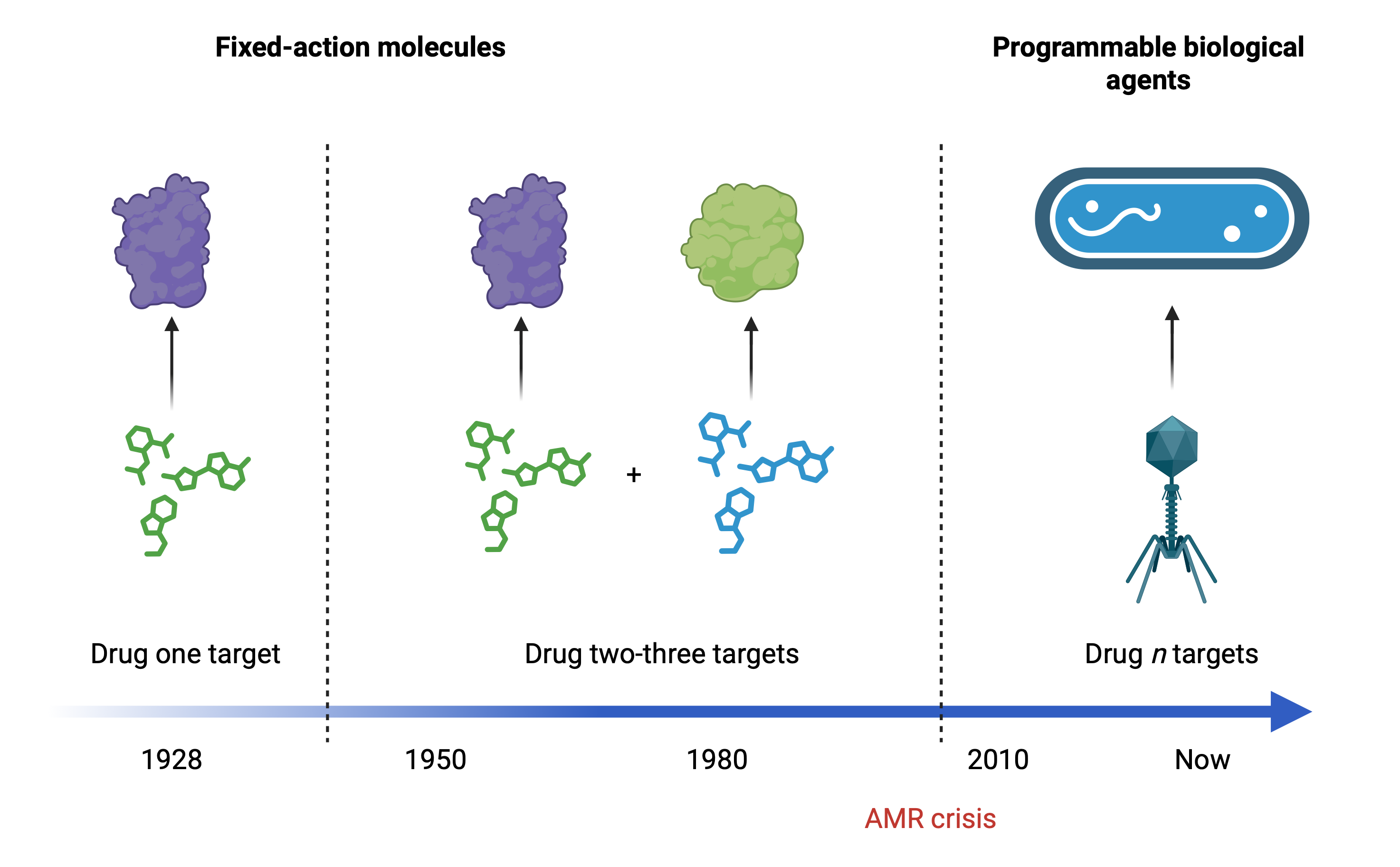
Viruses that infect bacteria, called bacteriophages or simply phages, have been locked in an evolutionary arms race with their hosts for billions of years. Every bacterial defense drives the emergence of new viral countermeasures. In our lab, we study this conflict at molecular scale. Our goal is to turn what we learn from it into new ways to fight antibiotic-resistant infections.
Understanding the rules of infection
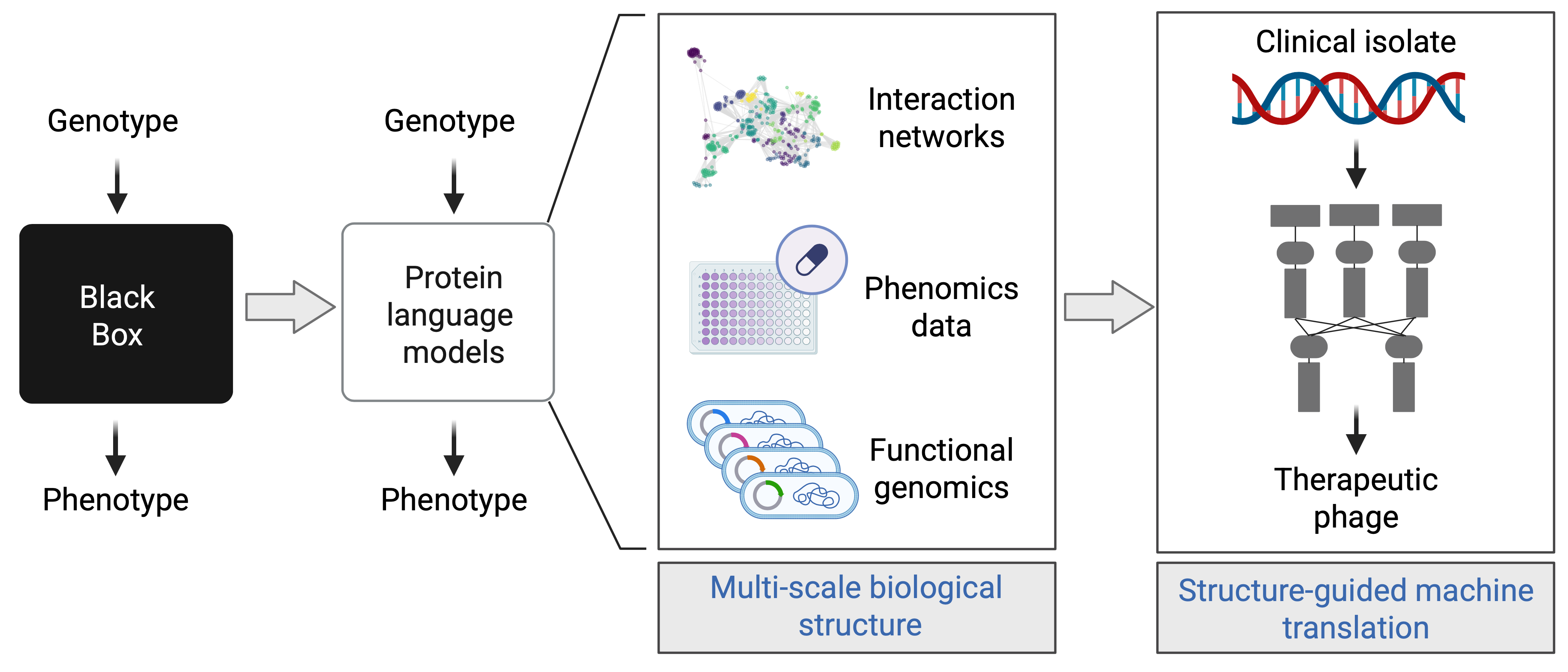
Phage infection triggers a rapid reorganization of bacterial machinery as hundreds of proteins interact, assemble and compete for control. We use interaction proteomics (large-scale mapping of protein–protein interactions) to characterize these molecular changes unfold inside infected cells. This lets us see how viruses rewire bacterial systems and which host complexes become targets.
By integrating these data with functional genomics and structural data, we identify which viral–host interfaces are essential for infection and which may serve as leverage points for therapeutic intervention.
Discovering bacterial defense systems
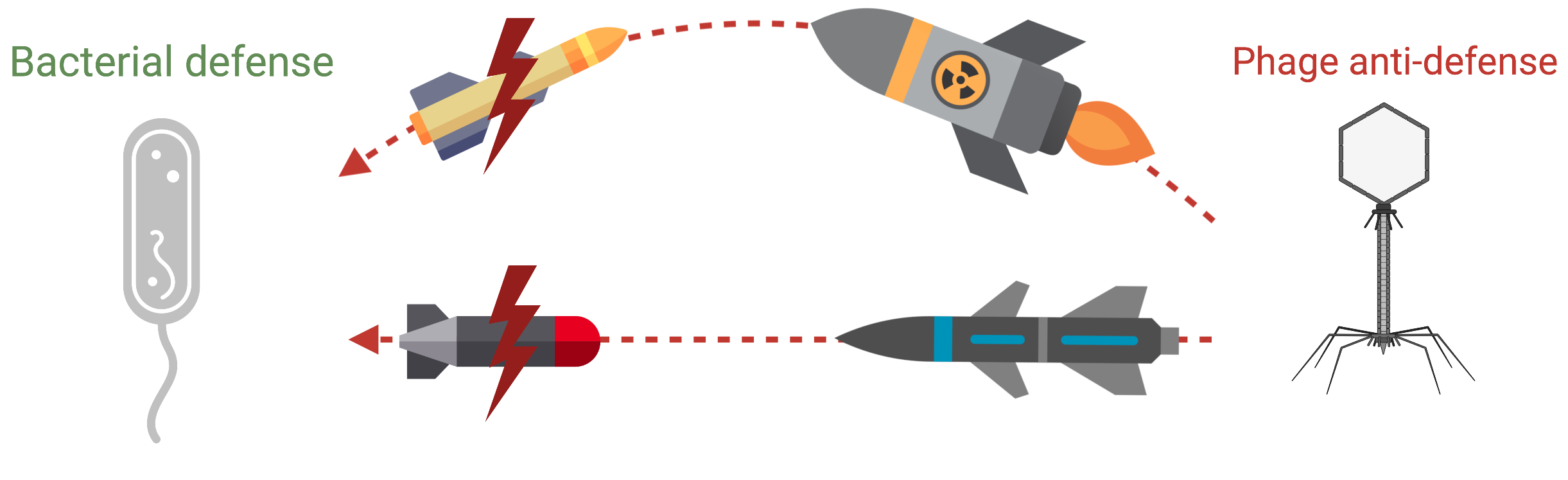
Just as humans have immune systems, bacteria have their own antiviral defense networks,including CRISPR and many others that are still undiscovered. We look for these “molecular alarm systems,” learn how they are triggered, and identify the phage proteins that disable them.
Understanding this tug-of-war helps us find new antibacterial strategies based on natural viral mechanisms.
Mapping the architecture of life
Most proteins act as part of larger assemblies rather than in isolation.
We use co-fractionation mass spectrometry (CF-MS) to identify and quantify these complexes directly from cells, capturing a snapshot of how they assemble, fall apart or swap partners during infection.
Our group also develops machine-learning methods to infer complex organization
and interaction networks from these data, providing a scalable framework for mapping proteome architecture.
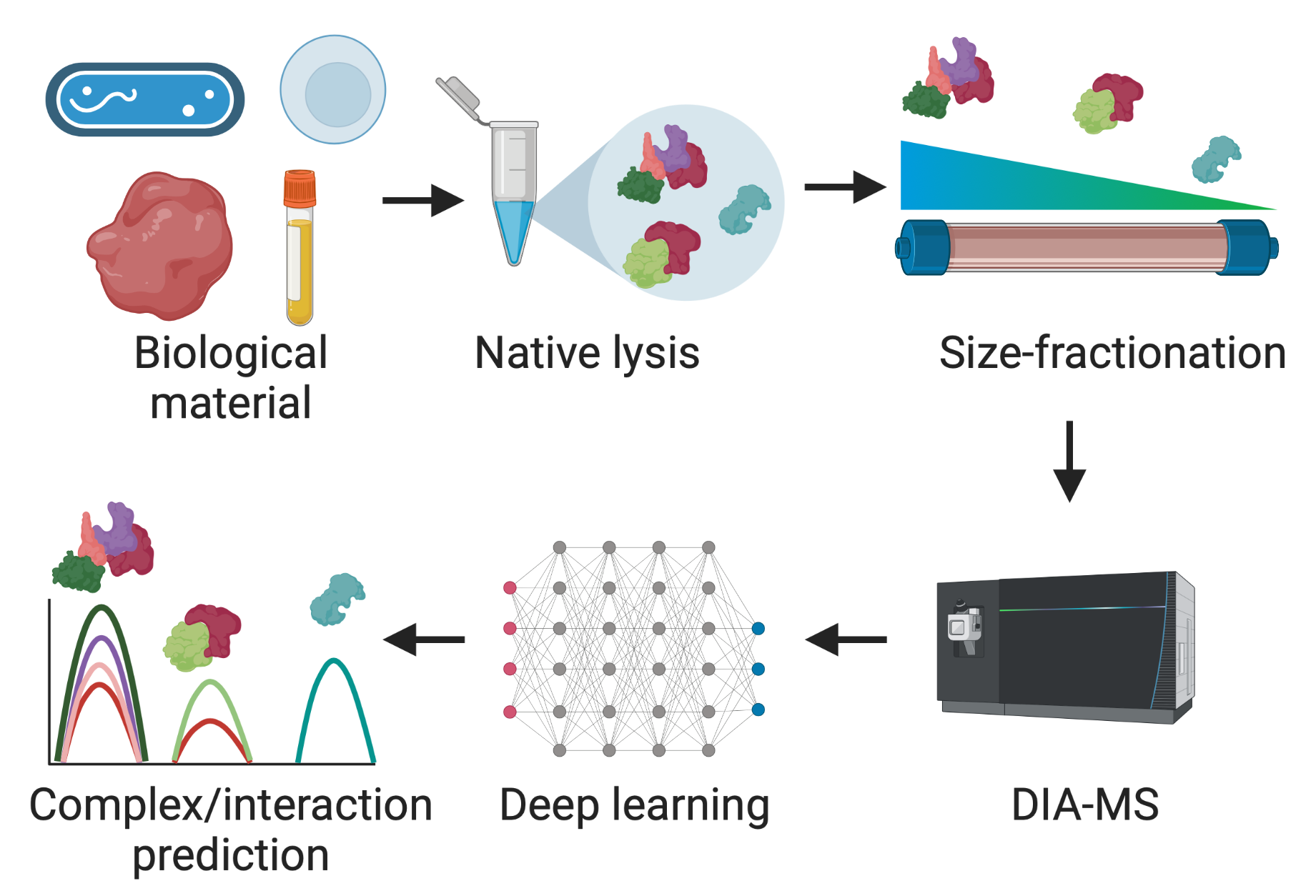 Schematic of a co-fractionation mass-spectrometry experiment.
Schematic of a co-fractionation mass-spectrometry experiment.
Designing phage-based therapeutics
By linking viral proteins to their bacterial targets, we can start to engineer phages and phage-derived molecules with defined functions: shutting down essential bacterial pathways, making pathogens more sensitive to antibiotics, or reshaping the course of infection.
This reflects a broader goal: to move from simply observing what viruses do to designing phages we can predict and control.
Interested in joining?
We welcome students and postdocs from biology, engineering and computational backgrounds who want to explore the viral frontier. Contact us to learn more.